
Molecular Oncology and Targeted Therapies
Overview
Genome instability is one of the hallmarks of cancer, affecting the initiation, evolution, and treatment response of the majority of cancers. To limit the accumulation of genomic alterations, cells have evolved complex mechanisms that coordinate cellular responses altogether known as the DNA Damage Response (DDR). However, in many cases the DDR is not sufficient to protect our genomes from either exogenous or endogenous insults, which may lead to cancer.
The overall goal of our laboratory is to increase our knowledge on the mechanisms that regulate the DDR and to identify novel therapeutic opportunities to treat or prevent cancer.
With this purpose, we utilize a wide range of approaches to investigate genome instability from the underlying molecular mechanisms to its ultimate consequences on health. We use cellular-based systems to perform screens, gain mechanistic insight and analyze cellular phenotypes in different genetic backgrounds. Besides, we generate genetically modified mouse models to address the physiological impact of specific genetic alterations and their influence on cancer. For both, cellular and mouse models-based studies, we use CRISPR/Cas9 technology to efficiently manipulate the genome. In collaboration with groups at the University of Copenhagen, we perform proteomic studies to investigate novel regulatory networks and the interplay between different proteins involved in the DDR. We also employ high content microscopy (HCM) as a routine approach to quantify alterations in the DDR, and to perform genetic and drug screens to identify novel therapeutic targets or compounds with clinical interest. The most relevant findings are validated and further characterized using mouse models.
Research highlights
In the past few years, we have investigated the role of PICH, a chromatin remodeler and DNA translocase essential for resolving ultrafine anaphase DNA bridges, in the progression of MYC-driven B-cell lymphomas. In our study published in Blood Cancer Journal (2024), we demonstrated that PICH deficiency limits the progression of MYC-induced lymphomas. Using a conditional PICH knockout mouse model, we demonstrated that targeting PICH could serve as a promising therapeutic strategy for MYC-driven lymphomas. These findings were further validated in several human Burkitt lymphoma cell lines. Given the potential interest of PICH as a therapeutic target in cancer, we are now focused on conducting compound screens to identify PICH inhibitors, which are currently unavailable.
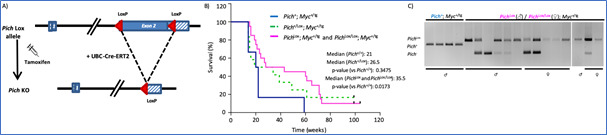
Figure 2. Pich deletion increases the survival of Eμ-Myc mice. A) Diagram of the Pich conditional KO allele and the expected Cre-mediated recombination. B) Survival of Eμ-Myc mice, without Pich deletion (blue line) and with Pich deletion in heterozygosity (green line) and homozygosity (pink line). Pich deletion was induced with tamoxifen injections from week 8 of age. C) Genotyping PCRs of the tumors at the humane endpoint. Of note, most tumors escaped from a complete Pich recombination, suggesting the relevance of PICH to sustain lymphoma progression.
We have recently published a comprehensive review about the known roles of the chromatin remodeler ATRX in regulating chromatin stability (Trends in Genetics, 2023). Given the central role of ATRX and the high frequency of ATRX mutations in human cancers, particularly brain tumors, we aim to deepen our understanding of the molecular mechanisms by which ATRX regulates chromatin stability, with the goal of identifying novel therapeutic opportunities. In this context, we have performed several drug screens to identify compounds that are synthetic lethal with ATRX deficiency, which we are currently validating in preclinical glioblastoma models.
During the past years, we have also investigated the role of FHIT in cancer, another gene frequently altered in cancer. Unlike other tumor suppressor genes, FHIT is not typically mutated; instead its entire locus is often lost due to its location within the highly unstable Common Fragile Site FRA3B. We have recently published a systematic analysis of FHIT gene alterations in cancer mining several cancer databases (Cell cycle, 2024). Our study suggests that FHIT loss is more likely a passenger event in cancer, in contrast to how its frequently described in the literature. Moreover, we are using a novel FHIT-deficient mouse model to elucidate the impact of FHIT loss in cancer and the molecular mechanism through which FHIT regulates genome stability and other pathways. As part of this research line, we have also conducted a compound screen to identify synthetic lethal interaction with FHIT loss. Although no compounds have yet been identified with synthetic lethality to FHIT, we discovered a novel cytidine analogue with potent anti-cancer activity in colon cancer cell lines. We are currently further characterizing and validating this compound in preclinical models.
Finally, we have performed CRISPR activation screens to identify genes whose overexpression may influence the response to therapies that induce replication stress. As an example, Figure 2 illustrates the use of hydroxyurea in these screens. We have identified genes of the DNA mismatch repair pathway and others such as ARID1A, which we are currently investigating.
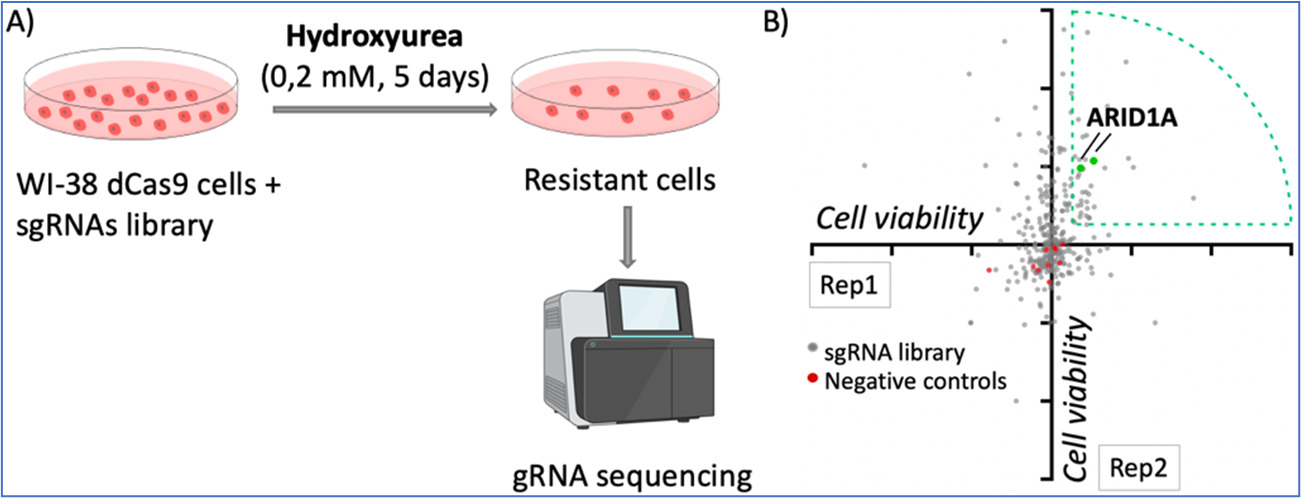
Figure 3. dCas9/CRISPR activation screening with hydroxyurea. A) Pipeline of the screening. WI-38 cells were infected with the dCas9/SAM system and a library of gRNAs targeting 68 tumor suppressor genes, with 3-5 gRNAS per gene. Cells were treated with low doses of hydroxyurea for 5 days and the resistant cells were analyzed by DNA sequencing to identify the representation of gRNAs. B) Diagram of gRNA enrichment of two replicates, each dot represents one gRNA. Two gRNAs targeting ARID1A were enriched in the two replicates performed.
Funding:
2021-2023: PY20-00755, Junta de Andalucía
2021-2024: PID2020-119329RB-I00, Ministerio de Ciencia e Innovación
2022-2026: Horizon-MSCA-2021-101072903, MSCA ITN network
If you are interested in joining our group as master student, PhD student or Postdoc, please send us your CV and motivation letter to: andres.lopez@cabimer.es
Recent publications:
Castejón-Griñán M, Albers E, Simón-Carrasco L, Aguilera P, Sbroggio M, Pladevall-Morera D, Ingham A, Lim E, Guillen-Benitez A, Pietrini E, Lisby M, Hickson ID, Lopez-Contreras AJ. PICH deficiency limits the progression of MYC-induced B-cell lymphoma. Blood Cancer J. 2024 Jan 23;14(1):16. doi: 10.1038/s41408-024-00979-y
Simón-Carrasco L, Pietrini E, López-Contreras AJ. Integrated analysis of FHIT gene alterations in cancer. Cell Cycle. 2024 Jan;23(1):92-113. doi: 10.1080/15384101.2024.2304509.
Aguilera P, López-Contreras AJ. ATRX, a guardian of chromatin. Trends Genet. 2023 Jun;39(6):505-519. doi: 10.1016/j.tig.2023.02.009.
Ruth KS, Day FR, Hussain J, …, Perry JRB. Genetic insights into biological mechanisms governing human ovarian ageing. Nature. 2021 Aug;596(7872):393-397. doi: 10.1038/s41586-021-03779-7.
Albers E, Avram A, Sbroggio M, Fernandez-Capetillo O, Lopez-Contreras AJ. Supraphysiological protection from replication stress does not extend mammalian lifespan. Aging (Albany NY). 2020 Apr 6;12(7):5612-5624. doi: 10.18632/aging.103039.
Atashpaz S, Samadi Shams S, Gonzalez JM, Sebestyén E, Arghavanifard N, Gnocchi A, Albers E, Minardi S, Faga G, Soffientini P, Allievi E, Cancila V, Bachi A, Fernández-Capetillo Ó, Tripodo C, Ferrari F, López-Contreras AJ, Costanzo V. ATR expands embryonic stem cell fate potential in response to replication stress. Elife. 2020 Mar 12;9:e54756. doi: 10.7554/eLife.54756.
Pladevall-Morera D, Munk S, Ingham A, Garribba L, Albers E, Liu Y, Olsen JV, Lopez-Contreras AJ. Proteomic characterization of chromosomal common fragile site (CFS)-associated proteins uncovers ATRX as a regulator of CFS stability. Nucleic Acids Res. 2019 Sep 5;47(15):8004-8018. doi: 10.1093/nar/gkz510.
Albers E, Sbroggiò M, Pladevall-Morera D, Bizard AH, Avram A, Gonzalez P, Martin-Gonzalez J, Hickson ID, Lopez-Contreras AJ. Loss of PICH Results in Chromosomal Instability, p53 Activation, and Embryonic Lethality. Cell Rep. 2018 Sep 18;24(12):3274-3284. doi: 10.1016/j.celrep.2018.08.071.
The complete and updated list of publications can be found at:
https://www.scopus.com/authid/detail.uri?authorId=8875970400

 Andrés Joaquín López Contreras
Andrés Joaquín López Contreras
- Dr. Lucía Simón Carrasco
- Dr. Paula Aguilera Aguilera
- Alba Guillén Benítez
- Manuel Luque Pérez
- Elena Pietrini
- Alba Soledad Rodríguez Raya



