e-mail: nestor.garcia@cabimer.es
Twitter:@NestorGarRod
ORCID ID: 0000-0002-4049-1604
Research Line:
- Understanding how cells sense and deal with DNA damage during replication as a critical barrier for carcinogenesis.

e-mail: nestor.garcia@cabimer.es
Twitter:@NestorGarRod
ORCID ID: 0000-0002-4049-1604
Research Line:

My research career has mainly focused on understanding how cells maintain the stability of their genome, specifically during the process of DNA replication. During the past years, I have made important contributions to the field of replicative stress. These contributions include the discovery of an unexpected role of RNA:DNA hybrids in origin-independent replication initiation within the ribosomal DNA locus (PNAS, 2015), or the finding that the DNA damage tolerance response occurs mostly at processed single-stranded DNA gaps that accumulate behind the replication forks in response to lesions on the DNA template (EMBO J, 2018; NAR, 2018; Mol Cell 2020; NAR 2024; Nature Comms. 2024.).
Recently, I have been awarded with an EMERGIA research grant (Junta de Andalucía), starting on May 2023, to stablish my own line of research on the mechanisms of DNA damage tolerance during replication in human cells, and the relevance of these mechanisms on human diseases such as cancer.

Previous positions:
DNA can be damaged by numerous endogenous and exogenous factors. To safeguard the genome against these insults, cells have evolved DNA damage checkpoints that sense the presence of damaged DNA, block cell cycle progression and ensure that DNA is fully repaired before resuming the cell cycle. However, in many cases, unrepaired lesions remain in the DNA when cells enter S-phase. In this scenario, cells employ DNA damage tolerance mechanisms to complete genome replication and prevent fork breakage (Figure 1). Importantly, these pathways are not restricted to the site of stalling but can also function behind the fork at single-stranded(ss)DNA gaps originated by re-priming of DNA synthesis downstream of lesions.
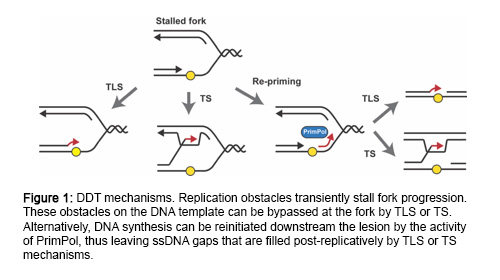
While it is well known that ssDNA is the signal that triggers the checkpoint response, it is less clear how and where ssDNA actually arises. Generally, it is assumed to accumulate at stalled replication forks by an uncoupling between replicative helicase and polymerase movement. However, recent evidence found in budding yeast support a model where, in response to polymerase-blocking lesions, replication forks do not stall but recover efficiently by re-priming downstream of the lesions, thus leaving ssDNA gaps behind the fork. Nucleolytic processing of such gaps is then required for efficient checkpoint activation. It is currently unknown whether this model also applies to vertebrate cells, and many questions remain regarding the cellular response to damaged DNA during replication. Where is the checkpoint signaling initiated in human cells, at the fork or behind the fork? What are the molecular mechanisms involved? Do nucleases play a role?
To address these and other questions, we will make use of a unique set of multidisciplinary approaches with the final goal to shed light on the molecular mechanisms of checkpoint activation and damage tolerance during replication of damaged DNA in human cells (Figure 2). An appropriate response to DNA replication stress acts as critical barrier to carcinogenesis. Thus, understanding the pertinent signaling pathways that deal with replicative stress is highly relevant to cell survival and human disease. Interestingly, recent evidence has identified ssDNA gaps as a cancer vulnerability. Thus, by manipulating how gaps are processed, we also hope to open new avenues in the treatment of cancer.
Projects as PI:
If you are interested in joining our research project as master or PhD student, please contact us at: nestor.garcia@cabimer.es
Articles:
A complete list of publications can be obtained at:



